Prof Nicolas Moitessier
McGill University
Objectives
In the present project, computational, organic and biological chemists team up in a feedback loop involving software development, computer-assisted drug design, modern chemical synthesis, and biochemical testing. Within this system, group members follow the lead compound from its moment of conception to its final performance in biochemical testing. The outcomes of this compound’s development are then reported to those members developing the software, and modifications or improvements in the software occur before the whole cycle begins again. The vested interest that the group shares for each prospective candidate helps to expedite the feedback process. We have decided to focus on two particular enzymes –Prolyl Oligopeptidase (POP) and Fibroblast Activation Protein-α– implicated primarily in cancer.
Drug Discovery and Development and Software Development
Although medicinal research has made tremendous advancements in the past decades, the expense and time associated with today’s drug design process is still overwhelming and the time it takes for a conceived lead compound to emerge as a marketed drug is well above a decade. Drug companies has invested large sums of money and time in high throughput screening (HTS), which still responds with low hit rates. Avoiding unproductive syntheses is a worthy goal not only in terms of cost and time efficiency but also environmental responsibility - as the amount of waste products from organic syntheses are greatly reduced. We believe a computational program that could provide us with highly accurate predictions of biological activity will be one solution to this critical problem.
At the heart of this medicinal chemistry project is our innovative computational platform, FORECASTER which seeks to discover new drugs or improve current drug design by accurately predicting the biological activity and other drug properties of proposed target compounds. We are developing this software in the hopes that in the future it can be used universally and applied to a large scope of physiological macromolecules, greatly improving efficiency in the world of drug design. The software we are inventing recognizes some of the most challenging elements of macromolecule structure and environment; molecular flexibility and solvation effects are two realistic aspects of chemical systems that are repeatedly ignored or greatly simplified in the use and programming of many other currently available modeling software.
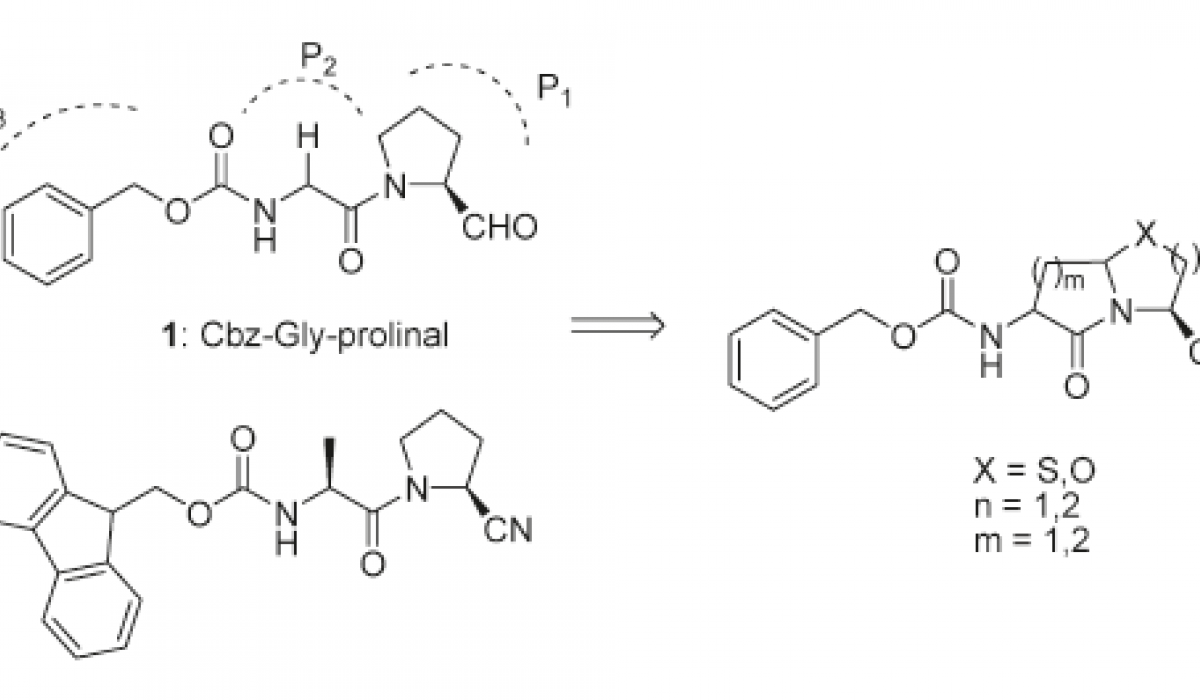
Drug Design and Synthesis
This software has been used to design novel inhibitors such as those in the Figure above.
Other Chemical series were designed and novel synthetic methodologies had to be developed and optimized. For example, a new expedient and green method to make bicylic scaffolds has been developed.
Our first generation of synthesized inhibitors has coincided with the preliminary and non automated version of our software; the next generation of compounds will be in response to the revised versions, and so on. The very first synthesized compounds are presently evaluated for their inhibitory potency. Throughout the project, there will be constant dialogue between the programmers and the medicinal chemists which will help to improve both the candidate compounds and the actual software being produced.
PRESENTED IN: